









Contenuti
Sindrome di Treacher-Collins
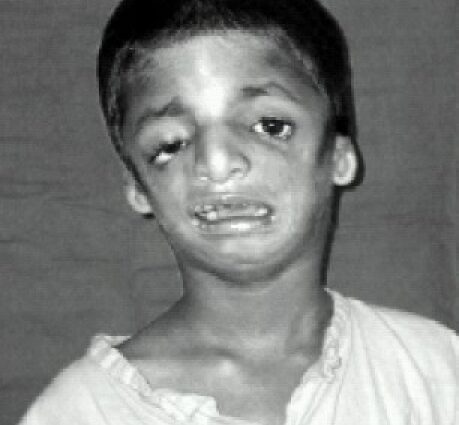
Una rara malattia genetica, la sindrome di Teacher-Collins è caratterizzata dallo sviluppo di difetti congeniti del cranio e del viso durante la vita embrionale, con conseguente deformità del viso, delle orecchie e degli occhi. Le conseguenze estetiche e funzionali sono più o meno gravi e alcuni casi richiedono numerosi interventi chirurgici. Tuttavia, nella maggior parte dei casi, la presa in carico consente di preservare una certa qualità della vita.
Cos'è la sindrome di Treacher-Collins?
Definizione
La sindrome di Treacher-Collins (dal nome di Edward Treacher Collins, che per primo la descrisse nel 1900) è una rara malattia congenita che si manifesta sin dalla nascita con malformazioni più o meno gravi della parte inferiore del corpo. viso, occhi e orecchie. Gli attacchi sono bilaterali e simmetrici.
Questa sindrome è anche chiamata sindrome di Franceschetti-Klein o disostosi mandibolo-facciale senza anomalie terminali.
Cause
Finora sono stati conosciuti tre geni per essere coinvolti in questa sindrome:
- il gene TCOF1, localizzato sul cromosoma 5,
- i geni POLR1C e POLR1D, localizzati rispettivamente sui cromosomi 6 e 13.
Questi geni dirigono la produzione di proteine che svolgono un ruolo chiave nello sviluppo embrionale delle strutture facciali. La loro alterazione attraverso mutazioni interrompe lo sviluppo delle strutture ossee (principalmente quelle della mascella inferiore e superiore e degli zigomi) e dei tessuti molli (muscoli e pelle) della parte inferiore del viso durante il secondo mese di gestazione. Sono interessati anche il padiglione auricolare, il condotto uditivo e le strutture dell'orecchio medio (ossicini e/o timpani).
Diagnostico
Le malformazioni facciali possono essere sospettate dall'ecografia del secondo trimestre di gravidanza, soprattutto in caso di malformazioni dell'orecchio significative. In questo caso, la diagnosi prenatale sarà stabilita da un team multidisciplinare a partire dalla risonanza magnetica (MRI) del feto, consentendo di visualizzare le malformazioni con maggiore precisione.
Il più delle volte, la diagnosi viene fatta da un esame fisico fatto alla nascita o subito dopo. A causa della grande variabilità delle malformazioni, deve essere confermato in un centro specializzato. Un test genetico su un campione di sangue può essere ordinato per cercare le anomalie genetiche coinvolte.
Alcune forme lievi passano inosservate o verranno rilevate fortuitamente in ritardo, ad esempio in seguito alla comparsa di un nuovo caso in famiglia.
Una volta effettuata la diagnosi, il bambino viene sottoposto a una serie di ulteriori esami:
- imaging facciale (raggi X, TAC e risonanza magnetica),
- esami dell'orecchio e test dell'udito,
- valutazione della vista,
- ricerca delle apnee notturne (polisonnografia)…
Le persone interessate
Si pensa che la sindrome di Treacher-Collins colpisca un neonato su 50, sia maschi che femmine. Si stima che ogni anno in Francia compaiano circa 000 nuovi casi.
Fattori di rischio
Si raccomanda la consulenza genetica in un centro di riferimento per valutare i rischi di trasmissione genetica.
Circa il 60% dei casi si presenta in isolamento: il bambino è il primo paziente della famiglia. Le malformazioni si verificano a seguito di un incidente genetico che ha colpito l'una o l'altra delle cellule riproduttive coinvolte nella fecondazione (mutazione “de novo”). Il gene mutato verrà poi trasmesso ai suoi discendenti, ma non vi è alcun rischio particolare per i suoi fratelli. Tuttavia, dovrebbe essere verificato se uno dei suoi genitori non sia effettivamente affetto da una forma minore della sindrome e non sia portatore della mutazione senza saperlo.
In altri casi, la malattia è ereditaria. Molto spesso, il rischio di trasmissione è uno su due per ogni gravidanza, ma a seconda delle mutazioni coinvolte, esistono altre modalità di trasmissione.
Sintomi della sindrome di Treacher-Collins
I tratti somatici delle persone colpite sono spesso caratteristici, con mento atrofizzato e sfuggente, zigomi inesistenti, occhi inclinati verso il basso verso le tempie, orecchie con padiglione piccolo e mal orlato, o addirittura del tutto assenti…
I sintomi principali sono legati a malformazioni della sfera ORL:
Difficoltà respiratorie
Molti bambini nascono con vie aeree superiori strette e bocche socchiuse, con una piccola cavità orale in gran parte ostruita dalla lingua. Di qui notevoli difficoltà respiratorie soprattutto nei neonati e nei lattanti, che si esprimono con russamento, apnee notturne e respiro troppo debole.
Difficoltà a mangiare
Nei neonati, l'allattamento al seno può essere ostacolato da difficoltà respiratorie e da anomalie del palato e del palato molle, talvolta sdoppiati. L'alimentazione è più facile dopo l'introduzione di cibi solidi, ma masticare può essere difficile e i problemi dentali sono comuni.
Sordità
Il disagio uditivo dovuto a malformazioni dell'orecchio esterno o medio è presente nel 30-50% dei casi.
Disturbi visivi
Un terzo dei bambini soffre di strabismo. Alcuni possono anche essere miopi, ipermetropi o astigmatici.
Difficoltà di apprendimento e comunicazione
La sindrome di Treacher-Collins non causa deficit cognitivo, ma sordità, problemi visivi, difficoltà di parola, le ripercussioni psicologiche della malattia nonché i disturbi indotti da cure mediche spesso molto pesanti possono causare un ritardo. lingua e difficoltà di comunicazione.
Trattamenti per la sindrome di Treacher-Collins
Cura infantile
Per facilitare la respirazione e l'alimentazione del bambino, a volte dalla nascita, può essere necessario un supporto alla respirazione e/o l'alimentazione tramite sondino. Quando l'assistenza respiratoria deve essere mantenuta nel tempo, si esegue una tracheotomia (piccola apertura nella trachea, al collo) per introdurre una cannula che assicuri direttamente il passaggio dell'aria nelle vie aeree.
Trattamento chirurgico delle malformazioni
Interventi chirurgici più o meno complessi e numerosi, relativi al palato molle, mascella, mento, orecchie, palpebre e naso possono essere proposti per facilitare il mangiare, la respirazione o l'udito, ma anche per ridurre l'impatto estetico delle malformazioni.
A titolo indicativo si chiudono le fessure del palato molle prima dei 6 mesi di età, i primi interventi cosmetici su palpebre e zigomi dai 2 anni, l'allungamento della mandibola (distrazione mandibolare) verso i 6 o 7 anni, riattacco di il padiglione auricolare intorno agli 8 anni, l'allargamento dei canali uditivi e/o la chirurgia degli ossicini intorno ai 10-12 anni... Altri interventi di chirurgia estetica possono ancora essere eseguiti nell'adolescenza...
Apparecchio acustico
Gli apparecchi acustici sono talvolta possibili dall'età di 3 o 4 mesi quando la sordità colpisce entrambe le orecchie. Sono disponibili diversi tipi di protesi a seconda della natura del danno, con una buona efficienza.
Follow-up medico e paramedico
Al fine di limitare e prevenire la disabilità, il monitoraggio periodico è multidisciplinare e coinvolge diversi specialisti:
- ORL (alto rischio di infezione)
- Oftalmologo (correzione dei disturbi visivi) e ortottista (riabilitazione oculare)
- Dentista e ortodontista
- Logopedista…
Spesso è necessario un supporto psicologico ed educativo.