









Contenuti
Sclerosi tuberosa di Bourneville
Che cos'è ?
La sclerosi tuberosa di Bourneville è una malattia genetica complessa caratterizzata dallo sviluppo di un tumore benigno (non canceroso) in diverse parti del corpo. Questi tumori possono quindi essere localizzati nella pelle, nel cervello, nei reni e in altri organi e tessuti. Questa patologia può anche causare seri problemi nello sviluppo dell'individuo. Tuttavia, le manifestazioni cliniche e la gravità della malattia variano da paziente a paziente.
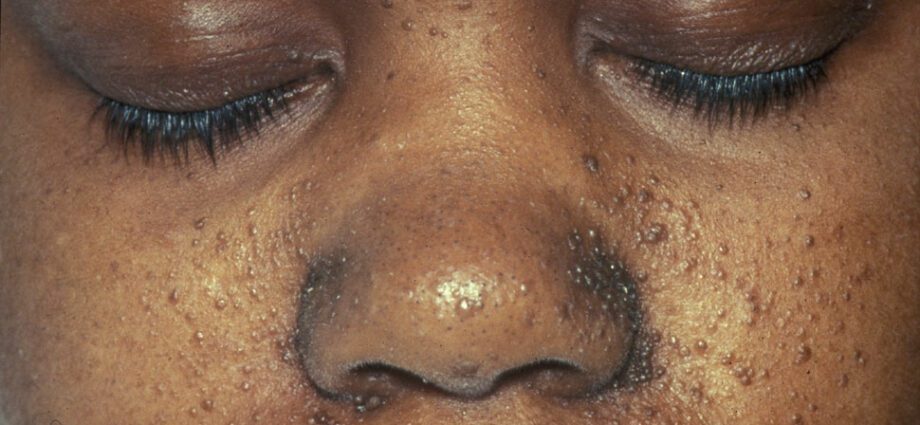
Le anomalie cutanee associate sono generalmente simili a macchie sulla pelle o ad aree in cui la pelle è più chiara rispetto al resto del corpo. Lo sviluppo di tumori in faccia è chiamato angiofibroma.
Nel contesto del danno cerebrale, i segni clinici sono crisi epilettiche, problemi comportamentali (iperattività, aggressività, disabilità intellettiva, problemi di apprendimento, ecc.). Alcuni bambini con la malattia hanno anche qualche forma di autismo, disturbi dello sviluppo, che influenzano le interazioni sociali e la comunicazione. I tumori cerebrali benigni possono anche causare complicazioni che possono essere fatali per il soggetto.
Lo sviluppo di tumori ai reni è comune nelle persone con sclerosi tuberosa. Ciò può causare gravi complicazioni nella funzione renale. Inoltre, i tumori possono svilupparsi nel cuore, nei polmoni e nella retina. (2)
È una malattia rara, la cui prevalenza (numero di casi in una data popolazione in un dato momento) è di 1/8 a 000/1 persone. (15)
Sintomi
Le manifestazioni cliniche associate alla sclerosi tuberosa di Bourneville variano a seconda degli organi colpiti. Inoltre, i sintomi associati alla malattia variano ampiamente da un individuo all'altro. Con sintomi che vanno da lievi a gravi.
I sintomi più ampiamente identificati di questa malattia includono crisi epilettiche, disturbi cognitivi e comportamentali, anomalie della pelle, ecc. Gli organi più colpiti sono: il cervello, il cuore, i reni, i polmoni e la pelle.
In questa malattia è possibile lo sviluppo di tumori maligni (cancerosi), ma sono rari e colpiscono principalmente i reni.
I segni clinici della malattia nel cervello hanno origine da attacchi a diversi livelli:
– danno ai tubercoli corticali;
– noduli ependimali (SEN);
– astrocitomi ependimali giganti.
Ne derivano: lo sviluppo di ritardo mentale, difficoltà di apprendimento, disturbi comportamentali, aggressività, disturbi dell'attenzione, iperattività, disturbi ossessivo-compulsivi, ecc.
Il danno renale è caratterizzato dallo sviluppo di cisti o angiomiolipomi. Questi possono portare a dolore ai reni e persino a insufficienza renale. Se si nota un forte sanguinamento, potrebbe essere dovuto a grave anemia o ipertensione. Possono essere visibili anche altre conseguenze più gravi ma rare, in particolare lo sviluppo di carcinomi (tumore delle cellule costituenti l'epitelio).
Il danno agli occhi può essere simile a macchie visibili sulla retina, causando disturbi visivi o addirittura cecità.
Le anomalie cutanee sono numerose:
– macule ipomelaniche: che provocano la comparsa di macchie chiare sulla pelle, in qualsiasi parte del corpo, a causa di una carenza di melanina, una proteina che dona colore alla pelle;
– la comparsa di macchie rosse sul viso;
– macchie scolorite sulla fronte;
– altre anomalie cutanee, dipendenti da individuo a individuo.
Le lesioni polmonari sono presenti in 1/3 dei pazienti con una leggera predominanza femminile. I sintomi associati sono quindi difficoltà respiratorie più o meno gravi.
Le origini della malattia
L'origine della malattia è genetica ed ereditaria.
La trasmissione coinvolge mutazioni nei geni TSC1 e TSC2. Questi geni di interesse entrano in gioco nella formazione delle proteine: hamartin e tuberina. Queste due proteine permettono, attraverso un gioco interattivo, di regolare la proliferazione cellulare.
I pazienti con la malattia nascono con almeno una copia mutata di questi geni in ciascuna delle loro cellule. Queste mutazioni limitano quindi la formazione di amartina o tubertina.
Nel contesto in cui le due copie del gene sono mutate, impediscono completamente la produzione di queste due proteine. Questa carenza proteica quindi non permette più all'organismo di regolare la crescita di alcune cellule e, in questo senso, porta allo sviluppo di cellule tumorali in diversi tessuti e/o organi.
Fattori di rischio
I fattori di rischio per lo sviluppo di una tale patologia sono genetici.
Infatti, la trasmissione della malattia è efficace attraverso una modalità autosomica dominante. In entrambi i casi, il gene mutato di interesse si trova su un cromosoma non sessuale. Inoltre, la presenza di una sola delle due copie del gene mutato è sufficiente per lo sviluppo della malattia.
In questo senso, un individuo che possiede uno di questi due genitori affetto dalla malattia ha un rischio del 50% di sviluppare lui stesso il fenotipo malato.
Prevenzione e trattamento
La diagnosi della malattia è prima di tutto differenziale. Si basa su criteri fisici atipici. Nella maggior parte dei casi, i primi segni caratteristici della malattia sono: la presenza di crisi epilettiche ricorrenti e ritardi nello sviluppo del soggetto. In altri casi, questi primi segni si traducono in macchie cutanee o nell'identificazione di un tumore al cuore.
A seguito di questa prima diagnosi, sono indispensabili ulteriori esami per convalidare o meno la diagnosi. Questi includono:
– una scansione cerebrale;
– una risonanza magnetica (Magnetic Resonance Imaging) del cervello;
– un'ecografia del cuore, del fegato e dei reni.
La diagnosi può essere efficace alla nascita del bambino. In caso contrario, è importante che venga eseguita il più rapidamente possibile per prendere in carico il paziente il prima possibile.
Attualmente, non esiste una cura per la malattia. I trattamenti associati sono quindi indipendenti dai sintomi presentati da ciascun individuo.
Di solito, vengono somministrati farmaci antiepilettici per limitare le convulsioni. Inoltre, vengono prescritti anche farmaci per il trattamento delle cellule tumorali del cervello e dei reni. Nel contesto dei problemi comportamentali, è necessario un trattamento specifico del bambino.
Il trattamento della malattia è solitamente a lungo termine. (1)